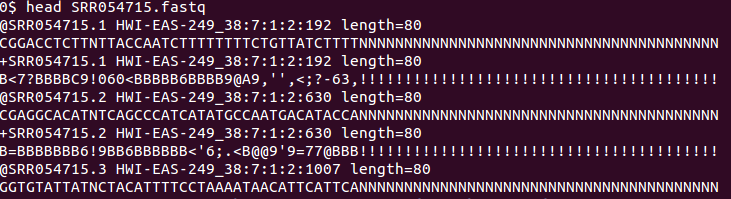
fastq格式与fasta格式相同的一点在于它们都是文本格式,不同之处在于前者提供了测序质量等更多的信息,而后者仅仅能提供序列(前者也提供序列)。
FASTQ format is a text-based format for storing both a biological sequence (usually nucleotide sequence) and its corresponding quality scores. Both the sequence letter and quality score are encoded with a single ASCII character for brevity. It was originally developed at the Wellcome Trust Sanger Institute to bundle a FASTA sequence and its quality data, but has recently become the de facto standard for storing the output of high throughput sequencing instruments such as the Illumina Genome Analyzer.[1]
sra格式则为二进制格式,由NCBI Short Read Archive提供,为高通量测序/下一代测序的“原始记录”。由于采用二进制方式进行压缩,该格式占用空间药效,可能限于经费的原因,NCBI SRA数据库现在仅提供了sra数据的下载。不过同时NCBI提供了SRA toolkit来进行sra格式->fastq格式(sam格式等)的转化。sra格式内部(貌似)以XML database的格式存储数据。
The Sequence Read Archive or Short Read Archive is a bioinformatics database and a collaboration between the European Bioinformatics Institute, the National Center for Biotechnology Information, and the DNA Data Bank of Japan. It provides a public repository for the “short reads” generated by High-throughput sequencing.
据笔者目测,一个约700M的sra文档,经fastq-dump转化后的大小在2.4G左右。
{kind=link}
fastq格式示例
{kind=link}
sra格式的内容 - 二进制文件