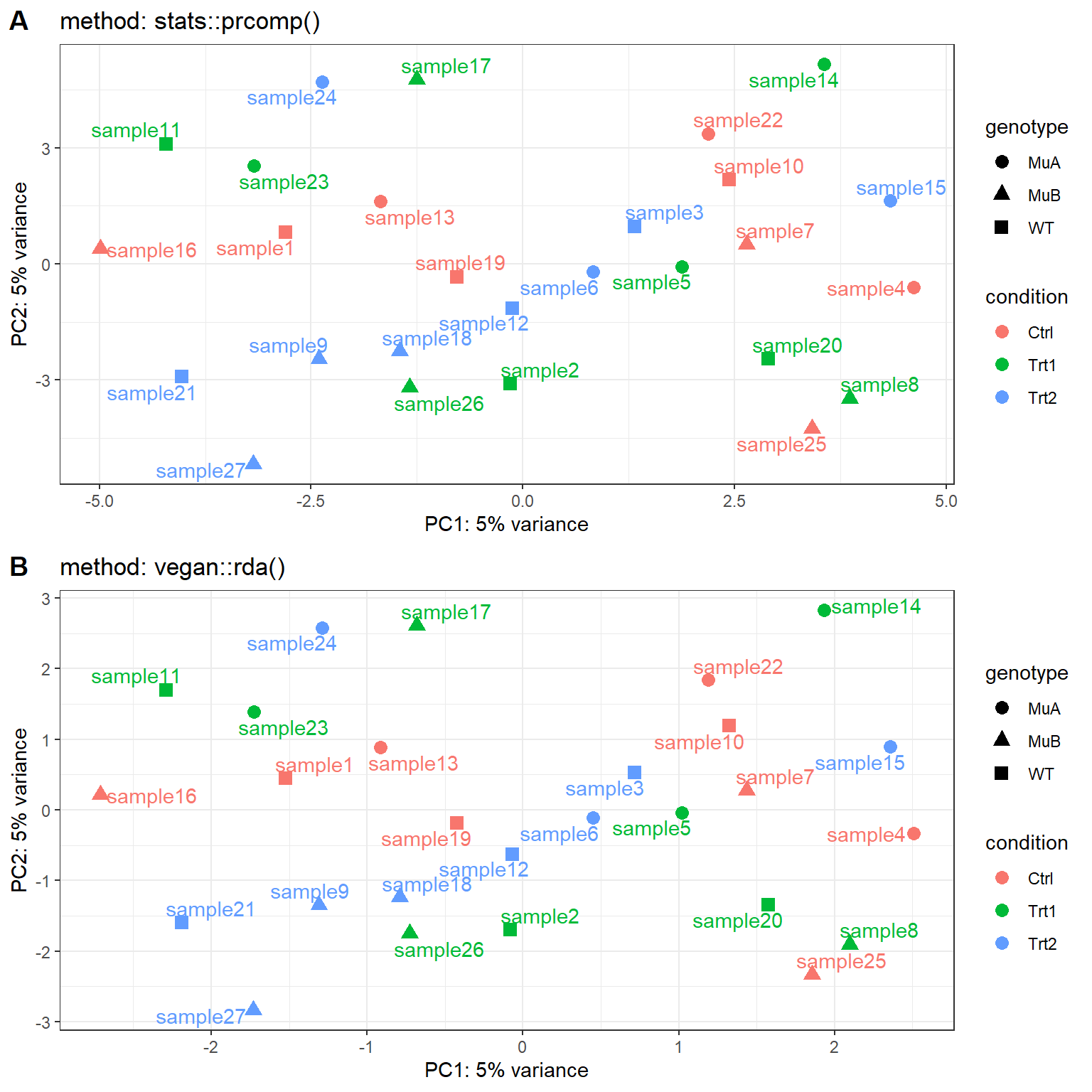
上次提及 PCA 分析的方法有很多种。那不同方法之间的得到的结果会有差异吗?
最近采用 PCA 分析 RNA-seq 样本之间的差异,得到了下面的结果。
生成示例数据
生成一个含有 1000 个基因, 27 个样品的数据集. 这 27 个样品来自于 3 个基因型(WT, Mutant1, Mutant2), 3 种处理(CK, Trt1, Trt2), 共分为 \(3 * 3 = 9\) 组, 每组 3 个重复, 合计 27 个样品.
library(DESeq2)
library(dplyr)
library(ggplot2)
theme_set(theme_bw())
dds <- makeExampleDESeqDataSet(n = 1000, m=27)
dds$condition <- factor(rep(rep(c("Ctrl","Trt1","Trt2"),3), 3))
dds$genotype <- factor(rep(rep(c("WT","MuA","MuB"), each=3),3))
# sample table
sample_table <- colData(dds) %>% as.data.frame() %>%
tibble::rownames_to_column(var="sample_id")
# 做 log2 变换
rld <- rlog(dds, blind = F)使用 stats::prcomp() 进行主成分分析
# 运行 PCA
pca <- stats::prcomp(t(assay(rld)))
# 计算解释度
percent_var <- pca$sdev^2/sum(pca$sdev^2)
# 绘图
df <- data.frame(PC1=pca$x[,1],
PC2=pca$x[,2], sample_id=colnames(rld)) %>%
left_join(sample_table)
mapping <- aes(PC1, PC2, color=condition, shape=genotype,label=sample_id)
p1 <- ggplot(df,mapping) +
geom_point(size=3) +
ggrepel::geom_text_repel(show.legend = F) +
xlab(paste0("PC1: ", round(percent_var[1] * 100), "% variance")) +
ylab(paste0("PC2: ", round(percent_var[2] * 100), "% variance")) +
ggtitle("method: stats::prcomp()")
p1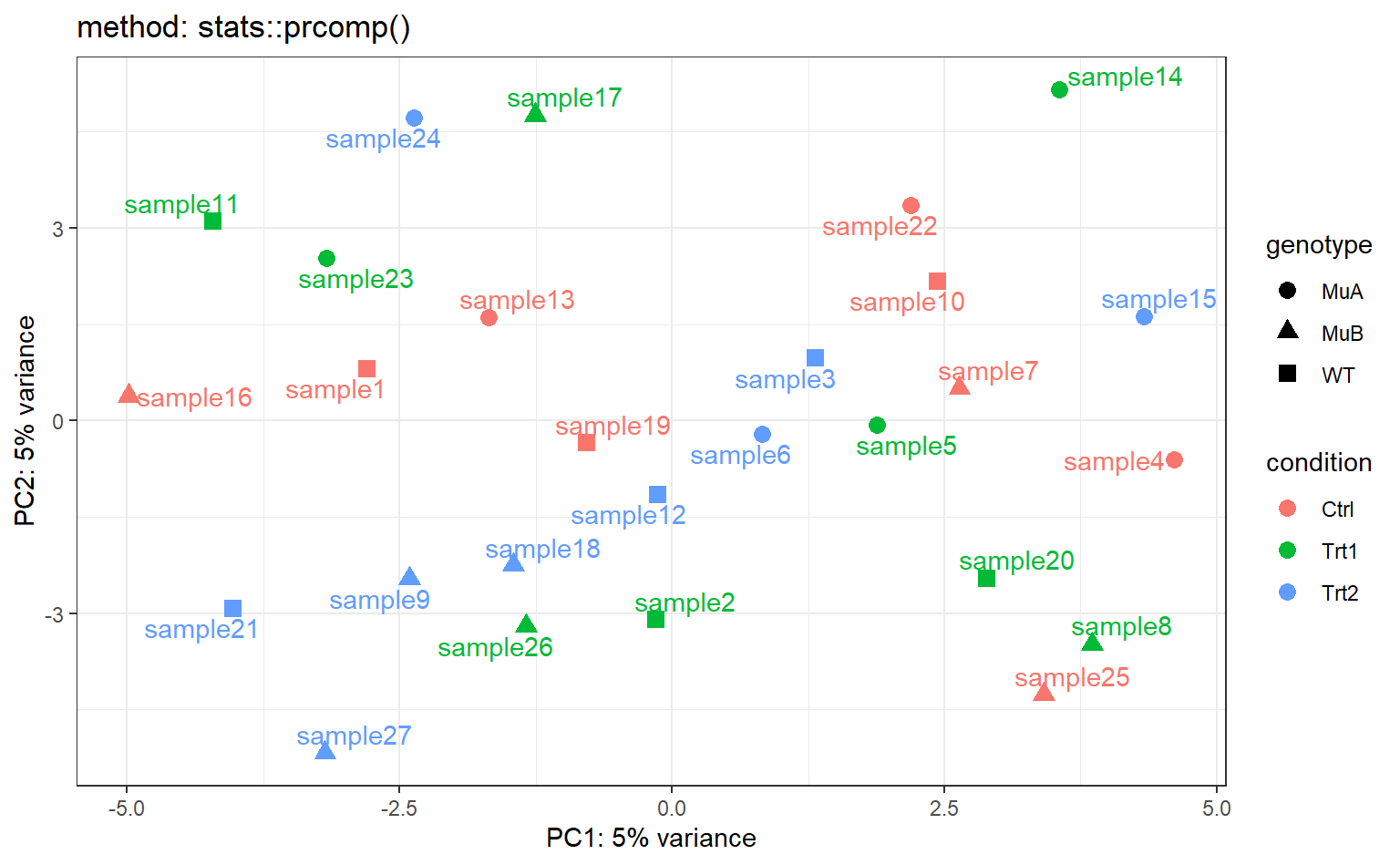
使用 vegan::rda() 做主成分分析
# rda() 分析
pca <- vegan::rda(t(assay(rld)))
# 计算解释度
percent_var <- pca$CA$eig/pca$tot.chi # rda() 的结果中信息比较完整
df <- vegan::scores(pca)$sites %>%
as.data.frame() %>%
tibble::rownames_to_column(var="sample_id") %>%
left_join(sample_table)
p2 <- ggplot(df,mapping) +
geom_point(size=3) +
ggrepel::geom_text_repel(show.legend = F) +
xlab(paste0("PC1: ", round(percent_var[1] * 100), "% variance")) +
ylab(paste0("PC2: ", round(percent_var[2] * 100), "% variance")) +
ggtitle("method: vegan::rda()")
p2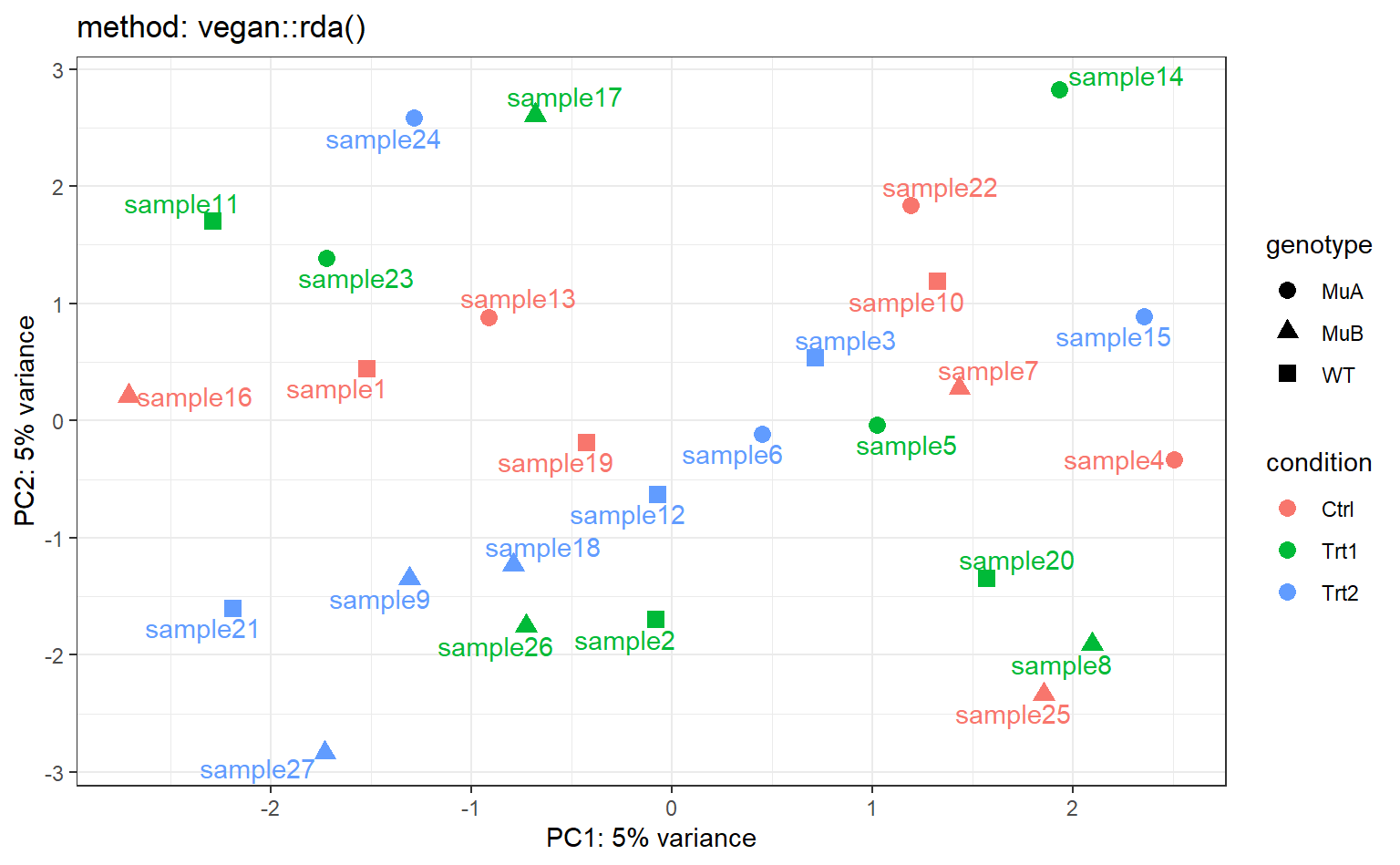
放在一起比较一下
可以看出,虽然两种方法计算的数值有差异,但是坐标位置是一致。
cowplot::plot_grid(p1,p2,labels = "AUTO",ncol=1,align = "hv")