GO term 太长, enrichplot 好局促怎么办?
例如下面这个样子:
# 可重复运行的代码拿去
library(clusterProfiler)
library(org.Hs.eg.db)
library(ggplot2)
data(geneList, package = "DOSE")
gene <- names(geneList)[abs(geneList)>3]
gene.df <- bitr(gene, fromType = "ENTREZID",
toType = c("ENSEMBL", "SYMBOL"),
OrgDb = org.Hs.eg.db)
ego <- enrichGO(gene = gene,
universe = names(geneList),
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
readable = TRUE)这个例子中, 有一个 “microtubule cytoskeleton …” 特别长, 事实上有些 GO term 比这还要长很多. 不巧在这个例子中没有出现, 那就拿它开刀吧.
dotplot(ego)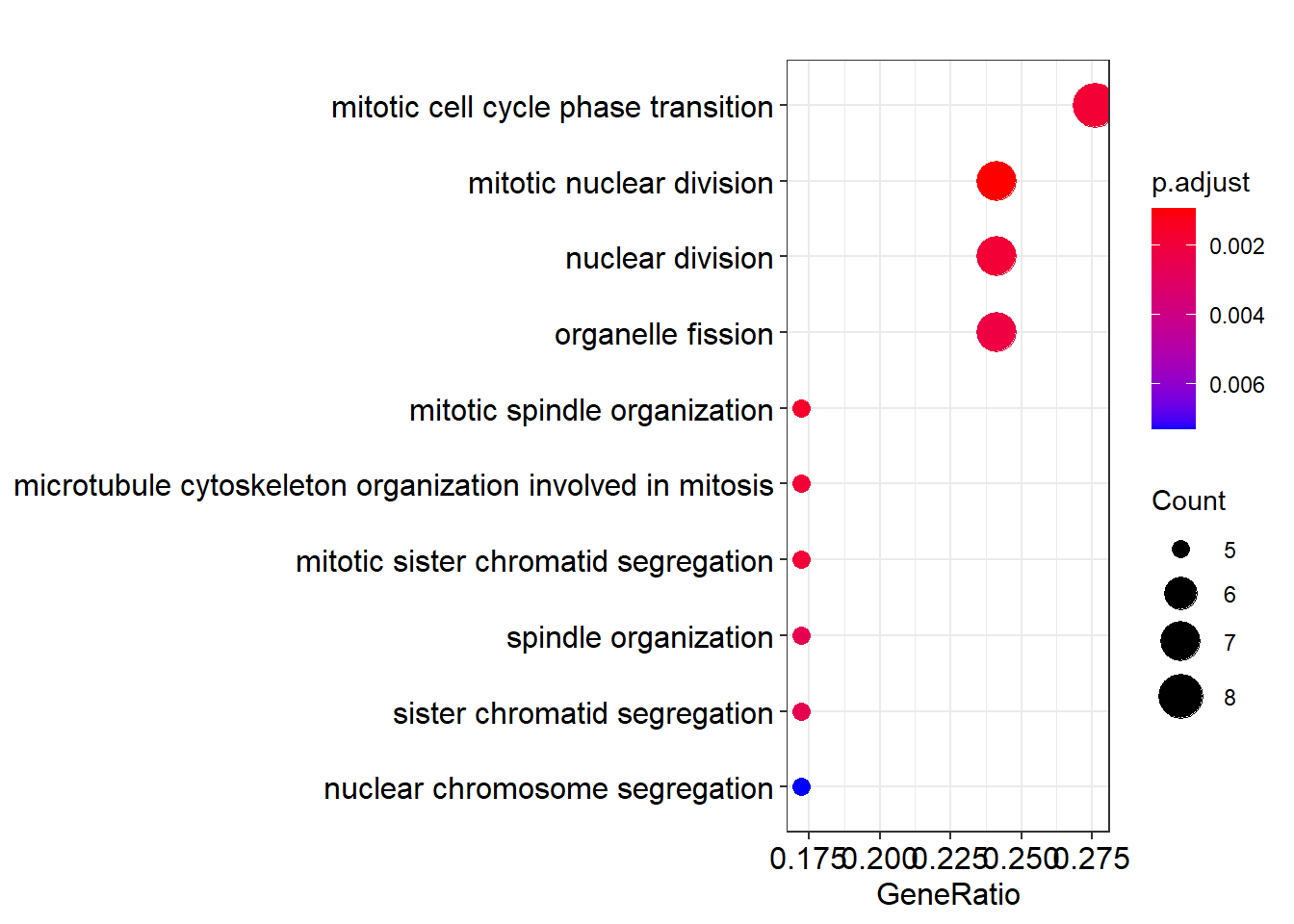
刀法比较犀利, 主要是定义一个短 label 的函数, 将其传递给 scale_y_discrete() 即可. 在输出ggplot 对象时做修改.
#' Truncate string vector of ggplot axis label
#'
#' @param label a ordered string vector
#' @param maxLen max length of character (nchar) to show in label
#' @param maxWord max count of words allowed to show in label
#' @param pattern Word separater
#' @param dot If true, three dots will added to truncated label
#'
#' @return a vector of truncated strings
#' @export
#'
#' @examples
short_label <- function(label, maxLen = 50, maxWord = 5, pattern = " ", dot = TRUE){
l <- strsplit(label, pattern)
short_label <- vector("character",length(l))
for (i in seq_along(l)){
truncated <- FALSE
s <- l[[i]]
if (length(s) > maxWord){
ll <- paste(s[1:maxWord], collapse = " ")
truncated <- TRUE
}
else{
ll <- paste(s, collapse = " ")
}
if (nchar(ll) > maxLen){
ll <- substr(ll, 1, maxLen)
truncated <- TRUE
}
if (dot & truncated) ll <- paste(ll, "...",sep = " ")
short_label[[i]] <- ll
}
attr(short_label, "pos") <- attr(label,"pos")
return(short_label)
}默认最多显示 50 个字符, 5 个单词.
dotplot(ego) + scale_y_discrete(label=short_label)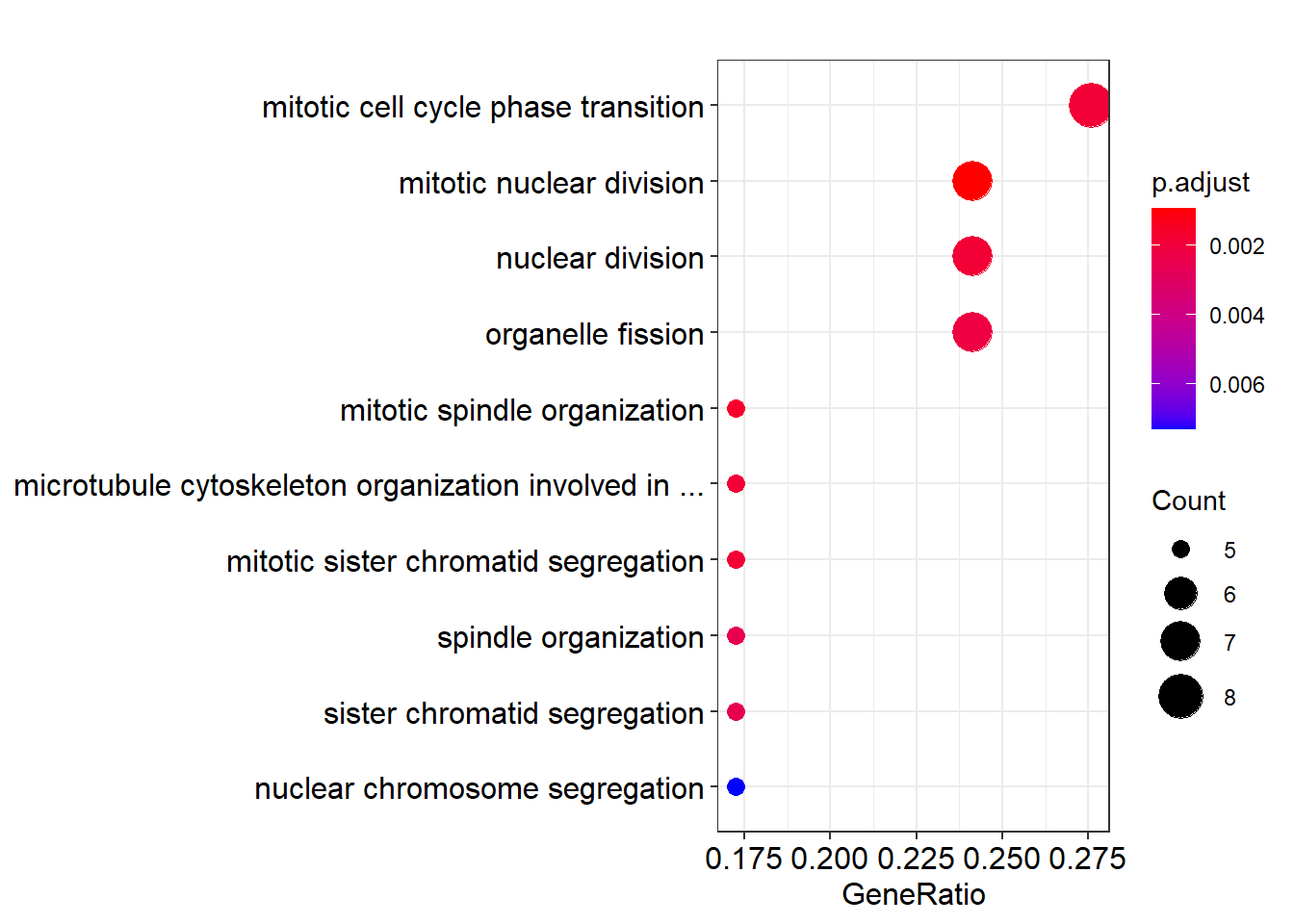
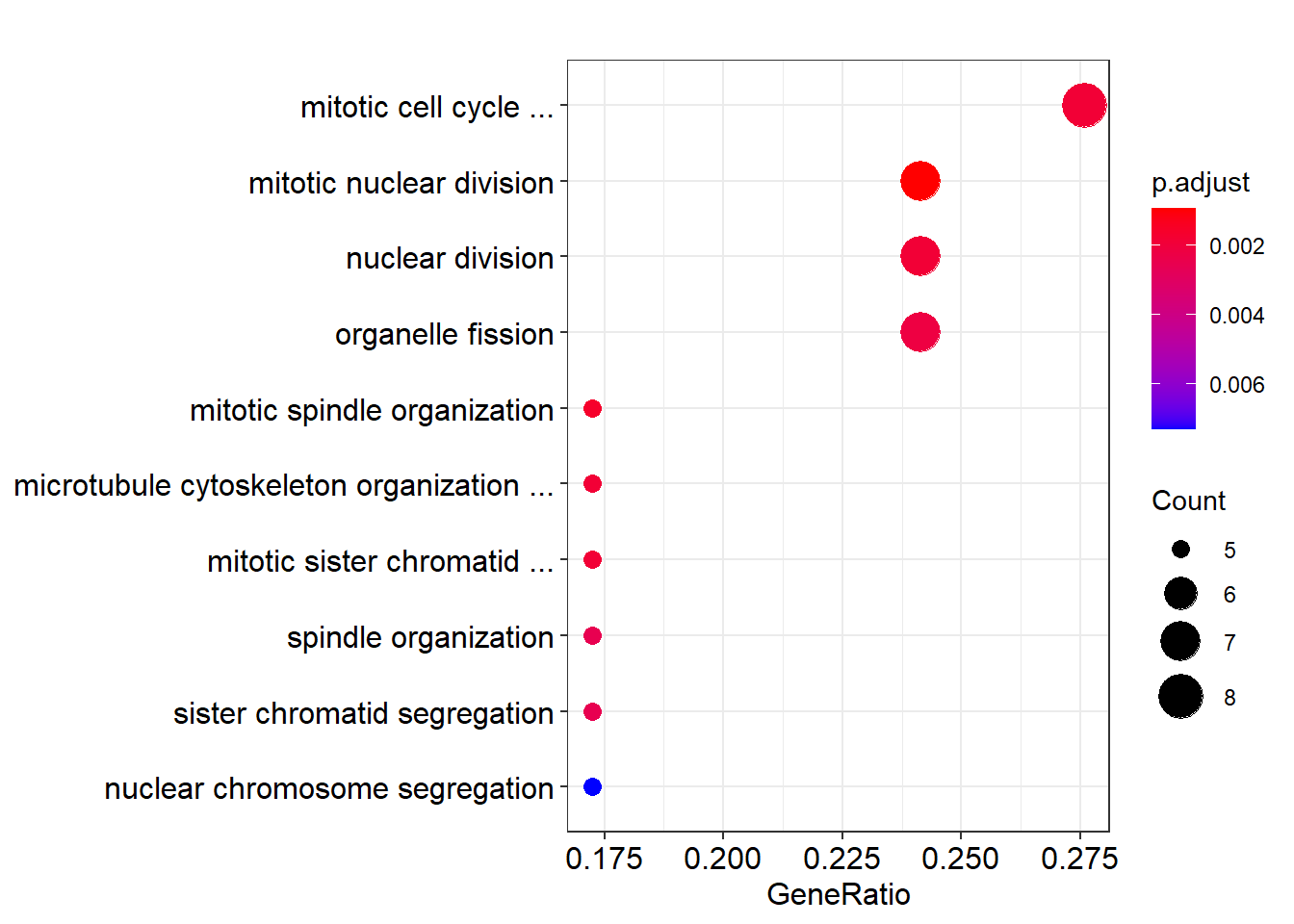
如果最多只显示 3 个单词, 则可以写成这样:
dotplot(ego) +
scale_y_discrete(
label=function(x)short_label(x,maxWord = 3)
)
注意: short_label() 带参数和不带参数时具有不同的调用方法.
不满意 enrichplot x轴排序怎么破?
我觉得 compareCluster 的输出可能会更加有比较价值. 但是发现 x 轴的排序有时候会有一些问题.
compareCluster 只能根据其本来固有的顺序排序, 这在程序上没有问题. 但用起来可能情况比较复杂.
data("gcSample")
names(gcSample)## [1] "X1" "X2" "X3" "X4" "X5" "X6" "X7" "X8"set.seed(0)
names(gcSample) <- sample(seq(0,28,by=4),8)
names(gcSample)## [1] "20" "0" "12" "24" "4" "16" "8" "28"ck <- compareCluster(geneClusters = gcSample, fun = "enrichKEGG")比如下面的例子中, 我们是想让 x 轴依数字大小(Time)排序的. 但是由于这个顺序与 list 顺序不同, 导致结果差强人意.
dotplot(ck)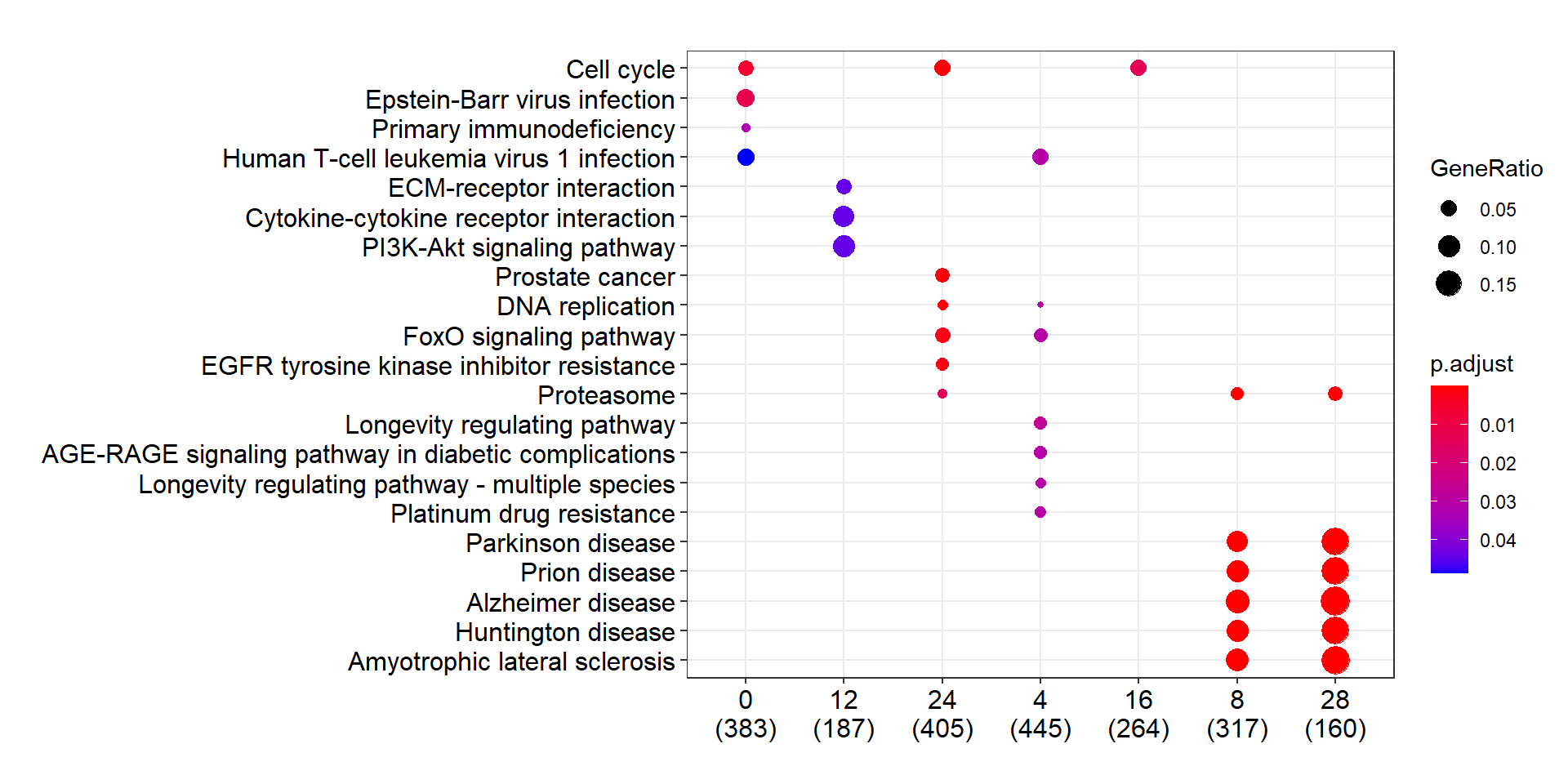
这只是一个 list 输入的例子, 可以通过改 name 变更顺序(就行我刚刚做的那样). 但是, 如果数据是放在 data.frame 中的, 存在多个分组条件, 每个条件按因子的 level 排好序.
那么由于 compareCluster 和 dotplot 操作时会将其转变成 character, 顺序会丢失, 导致最终结果是按照字母顺序输出的.
这种情况下, 你去改原始数据都是没有用的(需要给程序打补丁).
为了解决这个问题, 强行在 在已经输出的ggplot 对象上修改.
由于 ggplot 隐式输出的是一个对象, 其数据可以访问和修改. 出来的图, 先改一下再保存不就可以了吗?
p <- dotplot(ck)
levels(p$data$Cluster)## [1] "0\n(383)" "12\n(187)" "24\n(405)" "4\n(445)" "16\n(264)" "8\n(317)"
## [7] "28\n(160)"p$data$Cluster <- factor(p$data$Cluster,
levels = c("0\n(245)", "4\n(359)", "8\n(172)", "12\n(412)", "16\n(157)", "20\n(388)", "24\n(301)"),
labels = c(0,4,8,12,16,20,24))
p + xlab("Time (h)") + scale_y_discrete(label=short_label)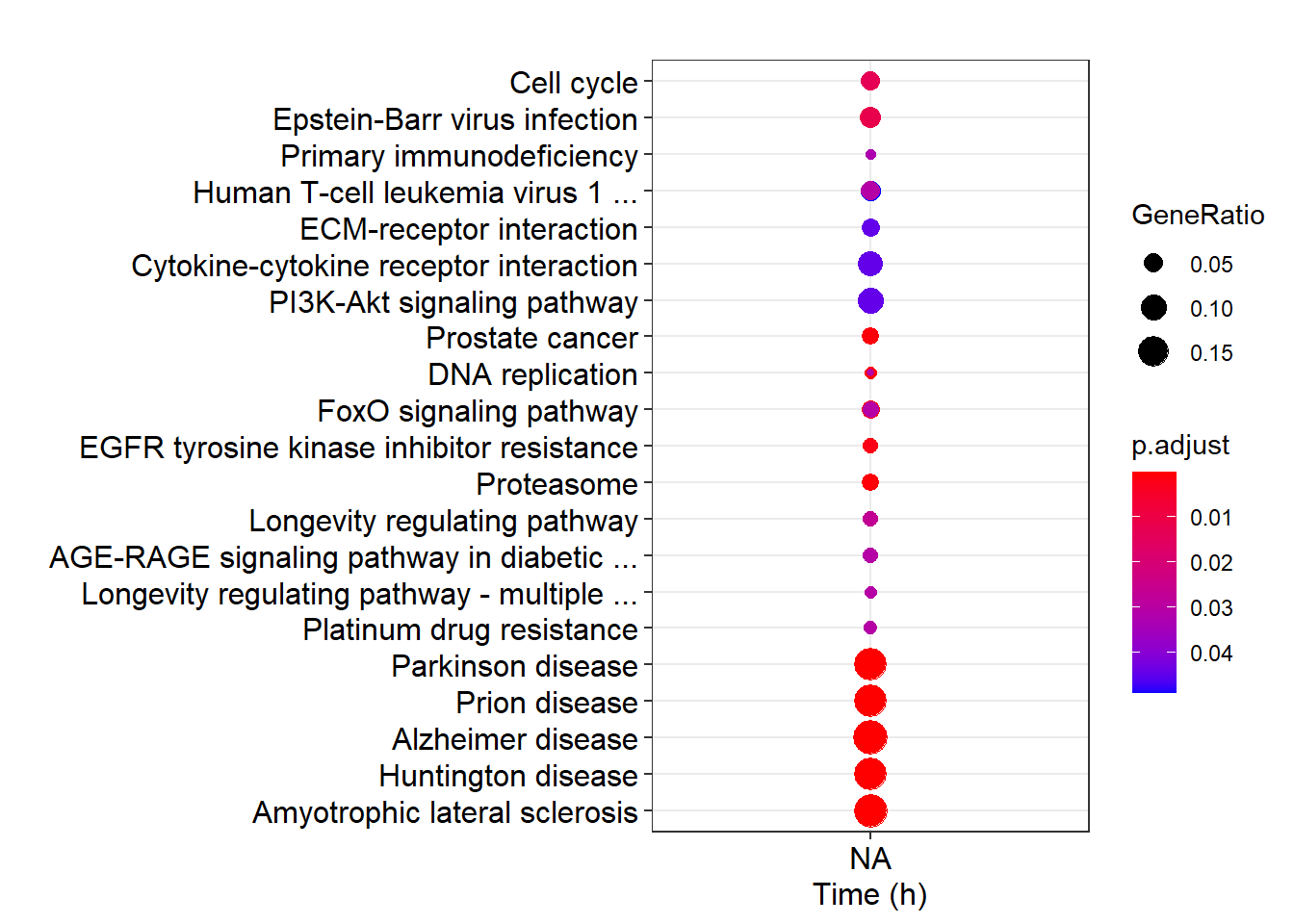
这两种“骚操作”, 分别相当于基因表达时“转录后修饰”和“翻译后修饰”. 嗯, 是有这么个意思. 都是在 “biobabble” 公众号学到的, 结合具体问题分享一下. 共同进步吧!