本文是我在华中农业大学JC Bioinformatics 2018暑期生物信息学培训班上的讲义
原核转录组
原核转录组课程讲授利用RNA-seq方法研究细菌基因组中基因表达差异的原理和方法。课程中不仅介绍了RNA-seq生物信息学分析的内容,还对RNA-seq的原理、文库构建等湿实验进行解读。
为了降低学习门槛,课程中的实例取自公开发表的细菌基因组,但是这不意味着相关原理和方法不能应用于其它微生物基因组。
另外,本课程涉及到的文库构建原理、方法、生物信息学工具软件在其它物种相关研究(各种基于高通量测序的研究方法)中也具有普适性。需要学生自己体会和进一步学习。
在开始本课程之前
你需要:
- 在你的电脑上已经安装了Linux双系统或虚拟机(推荐)
- Linux发行版推荐Ubuntu
- 虚拟机软件推荐VirtualBox
- 对Linux系统下的工作环境有一定认识
- 使用Bash或类似的命令行工具
- 了解命令参数的使用
- 会使用程序内嵌的帮助系统(help或man)
- 能够使用系统的包管理系统安装软件(如Ubuntu下的apt-get)
- 能够安装预编译好的生物信息学工具(包括设置PATH环境变量等)
- 在你的电脑上已经安装了最新版本的R
- 能够安装CRAN里面的包
- 能够安装Bioconductor里面的包
- 会使用R的命令行
- 会操作R的变量和数据
- 对ggplot作图有初步的认识
- 图形语法
- 能够使用ggplot2绘制简单的图形
- 在你的电脑上已经安装了最新版本的RStudio
- 已经初步认识了RStudio的界面
- 能够在RStudio中使用命令行
- 能够在RStudio中查看变量和输出
课程主要内容
- 转录组的原理:通过测序mRNA等的丰度得到基因表达情况
- RNA-seq的实验和分析流程
- 文库的构建:ssRNA-seq的介绍
- 短序列与基因组的比对
- 基因组注释信息在分析中的应用
- 差异表达基因的鉴定(差异基因数目上、表达量上的差异)
- 差异表达基因的富集分析
- 如何针对数据下一个结论
- 商业公司RNA-seq结果解析
- 文件夹的结构
- 结题报告(有用、无用、错误和正确的信息)
- 从头开始RNA-seq分析的流程
- Alignment -> Read Count
- 靠谱的基因表达差异工具:DESeq2
- 靠谱的基因富集分析工具:ClusterProfiler
- 案例研究:重现论文中的数据图表
- 研究目的:BS、PP对VP基因表达的影响
- 实验设计:共培养和单独培养的RNA-seq
- RNA-seq数据分析
- 结论
转录组的原理:通过测序mRNA的丰度得到基因表达情况
前一段时间,有一个非常火的电视剧:三国机密之潜龙在渊。讲述了汉室与曹操之间纠葛的关系。 里面有曹丕与曹植争宠。让我想到了他们还有个弟弟:曹冲。历史总是很有意思的。 曹冲十三岁就死了,让人遗憾。但是,曹冲称象的故事广为流传。

古时候没有那么大量程的秤,采用了化整为零的方法。所以有一头大象 = 一堆石头的换算。
那如果来了一群大象,就可以分别用不同的一堆石头来定量,得出大象之间的重量差异。
- 大象A = 石头堆A
- 大象B = 石头堆B
- 大象C = 石头堆C
- ……
这其实跟RNA-seq检测基因表达差异的情况非常类似。
我们知道中心法则:DNA编码的基因通过转录变成了RNA(其中大量是rRNA)。RNA-seq就是通过比较RNA的丰度来定量基因表达的差异。
问题:
- 基因组DNA的长度范围是什么?
- RNA的长度范围是什么?
- 测序的长度是什么?
因为测序的读长(人类和机器的能力)远低于DNA或RNA的长度(自然的能力),所以我们不得不采用曹冲称象的方法来还原基因组和转录组。
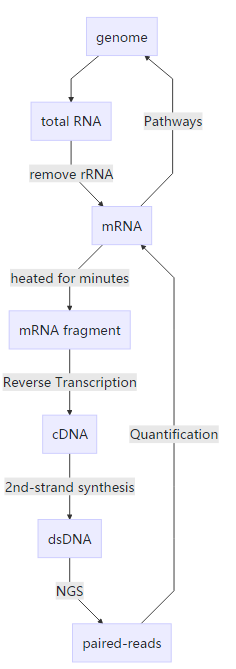
建测序文库是将RNA打断成小片段的过程。测序之后,得到一堆一堆的小石头(称为Reads,长度100-150bp)。
生物信息学分析是将小石头对应到基因表达上的过程,是建库的逆过程。通过比较各个基因石头堆中Reads的数目,可以得出基因表达差异。
RNA-seq的实验和分析流程
RNA-seq实验一般至少有两组样品,分别是对照组和实验组,每组一般设3个重复。
| 对照组 | 实验组 | 实验设计 |
|---|---|---|
| wild type | mutant of a transcriptional regulator | 转录因子调控的靶基因 |
| non-treated | treated | 处理引起(诱导)的基因表达变化情况 |
| 0 h | 6h, 12h, etc | 基因表达随时间的变化情况 |
实验设置重复对于发表论文是非常重要的。没有重复的结果很难被杂志和审稿人接受。
一个成功的RNA-seq实验,合理的实验设计是基本需要。获得实验中所需的样品可能要花很多精力。另外,由于RNA不稳定的特性,样本制备和保存也要特别注意。
测序一般由商业公司完成。剩下的主要工作就是生物信息学分析。
注意:这一部分先讲基本原理,下一部分讲生物信息学操作。
文库的构建:ssRNA-seq的介绍
目前主流的RNA-seq文库构建方法分为链特异性和非特异性文库。链特异性文库包含了RNA所在DNA链的信息,成本只比后者略高,因此建议构建链特异性文库(Strand-specific RNA-seq,ssRNA-seq)。
我们知道,DNA是双链,RNA是单链。所以同样一段DNA可以有两种序列不同的RNA产生。ssRNA-seq在建库的过程中对序列标记,保留了链特异性信息。
常见的链特异性文库构建方法是dUTP方法。
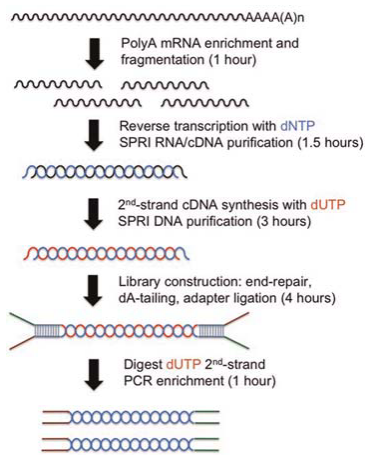
在合成第二链的时候,使用dUTP替代dTTP,合成之后通过UDG酶降解第二链,从而得到特异性的文库。
参考文献:
- Zhong S et al. (2011). High-Throughput Illumina Strand-Specific RNA Sequencing Library Preparation. Cold Spring Harb Protoc 2011: pdb.prot5652.
- Levin JZ et al. (2010). Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Meth 7: 709–715.
- Zhang Z et al. (2012). Strand-specific libraries for high throughput RNA sequencing (RNA-Seq) prepared without poly(A) selection. Silence 3: 9.
短序列与基因组的比对
我们前面提到,每个基因(大象)测序(称量)之后都对应一堆Reads(石头)。但问题是,这些石头是一大堆。如何将它们分开呢?
这个过程就是短序列比对。
短序列比对需要将百万量级的Reads快速、准确的比对到基因组上。采用了不同于BLAST的算法、结果呈现方式等。
基因组注释和差异表达基因的鉴定
在上一步将reads比对到基因组上后,可以根据基因组注释信息确定对应基因上序列的个数。
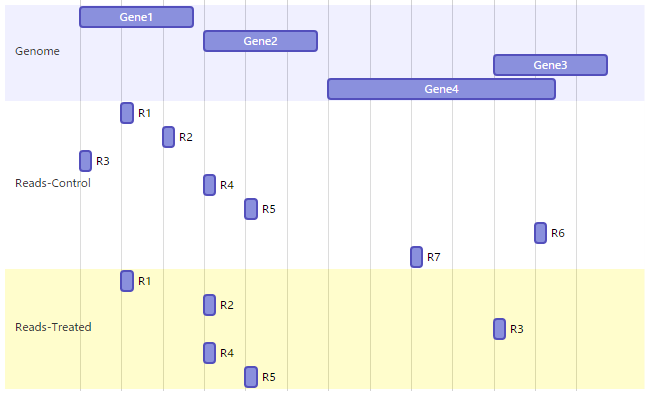
简单说来,会有下面的结果。
| Gene | Control | Treated | Fold Change |
|---|---|---|---|
| Gene1 | 3 | 1 | 1/3 |
| Gene2 | 2 | 3 | 3/2 |
| Gene3 | 1 | 1 | 1/1 |
| Gene4 | 2 | 1 | 1/2 |
当然,事实上的情况比这要复杂。上图中,Control一共有7个Reads,而Treated一共有5个Reads;Gene4的长度也比其它要长;Gene3和Gene4还有一部分重叠。这些情况在真正的分析时都要考虑,会使用相应的模型和统计算法来完成。
好消息是,生物信息学软件将为你做好了这一切。所以,你不要太担心。
差异表达基因的富集分析
在上一步之后,我们将会得到差异表达的基因。这些基因可能会有很多,表达量变化幅度可能也不一样。比如,我们分析发现:200 个基因表达上调,150 个基因表达下调。这有什么生物学意义呢?
这就是富集分析要解决的问题。富集分析可以告诉你,基因表达变化主要发生在哪些代谢途径?哪个细胞结构(细胞壁、细胞膜、细胞器)?
假如基因组中一共有2000个基因,其中200个是与氨基酸代谢相关的(酶、调控蛋白等),占全部基因的十分之一。而恰好200个上调表达的基因里面,100个都是氨基酸代谢相关的基因。那意味着什么?
如果基因表达差异是随机的,那我随机选择200个基因,按照十分之一的几率,其中最有可能是有20个左右的氨基酸代谢相关基因。但是在200个上调表达的基因中,却有一半的基因是氨基酸代谢相关基因。说明氨基酸代谢途径是显著受影响的代谢途径,在差异表达基因基因中富集。这就是富集分析。
富集程度以超几何分布概率公式计算。其中N是基因总数,m是具某一属性(如氨基酸代谢相关)的基因总数(m < N)。n是总的差异表达基因,k是具同一属性(如氨基酸代谢相关)基因的个数。
$$f(k;n,m,N) = {{{m \choose k} {{N-m} \choose {n-k}}}\over {N \choose n}}$$
在上述的例子中,计算phyper(100,1800,200,200)的值为$3.0 \times 10^{-56}$。如果出现这样的结果,肯定不是一个巧合。
如何针对数据下一个结论
小概率事件不可能发生的,发生了小概率事件,那只能说明处理组中氨基酸代谢相关途径显著上调了。意味着细胞开始大量代谢氨基酸。是合成?还是降解?都可以用类似的富集分析方法去分析。
这就是RNA-seq的结论部分。
商业公司RNA-seq结果解析
测序公司都会出具一个结题报告,并将分析结果打包给你。
基本上,上面描述的分析,测序公司都会包含在内(只多不少)。
但是测序公司的结题报告,存在一些不足之处。
- 千篇一律,有用的信息夹杂在无用的信息之间;
- 做得好测序,却做不好数据分析和解读;基本上,越到结题报告的后面,越让人看不下去。
- 公司的分析数据库常常缺乏更新;
- 你不能设定分析的参数;
- 甚至包含着错误的信息。
结题报告(有用、无用、错误和正确的信息)
下面是某大基因公司的结题报告。
略
下面是某诺公司的结题报告。
略
一般来讲,共有类似的文件夹结构。
文件夹的结构
--Results
+- Rawdata | CleanDataAssess
+- Genome
+- Map | MappingAssess
+- Expression | QuantitativeAnalysis
+- DESeq
+- Structure | StructureAnalysis
report.pdf
...
你需要什么内容,应该可以按图索骥了。
从头开始 RNA-seq 分析
将 raw reads 比对到参考基因组
使用的生物信息学工具/软件:bowtie2或hisat2或其它短序列比对软件。
Bowtie 2 is an ultrafast and memory-efficient tool for aligning sequencing reads to long reference sequences. HISAT2 is a fast and sensitive alignment program for mapping next-generation sequencing reads (whole-genome, transcriptome, and exome sequencing data) against the general human population (as well as against a single reference genome).
测序结果是以 fastq格式分两个Read保存的,通常会压缩,扩展名推荐为 *.fastq.gz。
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
现在的测序结果一般是双端测序,会有两个分开的文件。
短序列比对虽然与常用的BLAST比对差别较大,但是二者实际上做的是同样的工作:都是把一段较短的序列比对到数据库中较长的序列上。
首先需要建立一个基因组数据库索引,然后才能把测序得出的Paired-reads(fastq格式文件)比对到基因组上,比对结果输出为*.sam格式文件。
关于
fastq格式,参考:FASTQ格式
关于
*.sam格式,参考:SAM格式说明。
## 从基因组序列 genome.fasta 创建名为 genome 的 hisat2 专用索引
hisat2-build genome.fasta genome
## 运行下列命令将 forward.fq.gz 和 reverse.fq.gz 中的测序结果比对到 genome 索引,文件输出为 file.sam
hisat2 -1 forward.fq.gz -2 reverse.fq.gz -x genome -S file.sam
file.sam是TAB分隔的文本文件,可以用less file.sam查看。
计算落到每个基因上面的 Reads 个数 - Reads count
使用的生物信息学软件有:samtools,htseq,NCBI数据库。
Samtools is a set of utilities that manipulate alignments in the BAM format. It imports from and exports to the SAM (Sequence Alignment/Map) format, does sorting, merging and indexing, and allows to retrieve reads in any regions swiftly.
HTSeq is a Python package that provides infrastructure to process data from high-throughput sequencing assays.
下图是一个计数模型。
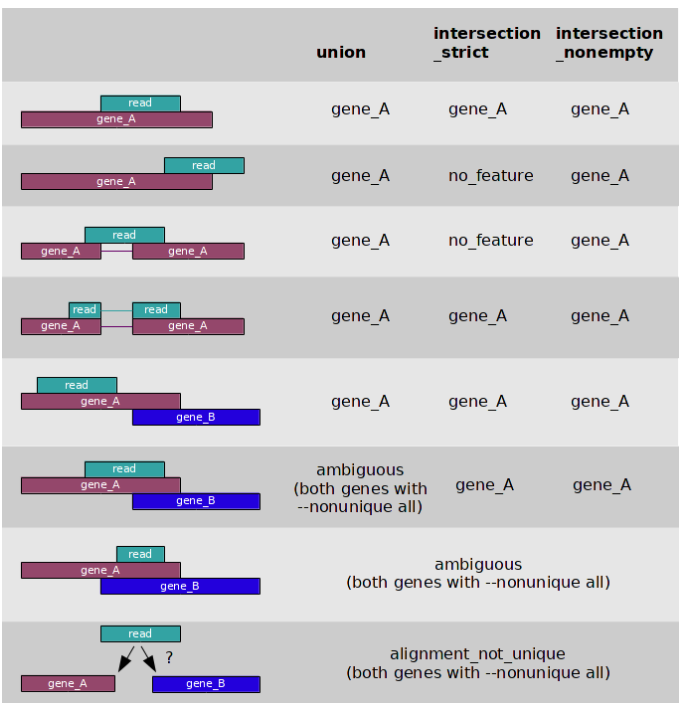
## 格式转换 SAM -> BAM
samtools view -bS file.sam -o file.bam
## 将比对结果按照 read 的名称排序
samtools sort -n file.bam #sort bam by name
## 计算落到各个基因上面的 Reads 个数
htseq-count -f bam --stranded=yes --type=CDS --idattr=id file.bam gene.gff
靠谱的基因表达差异工具:DESeq2
能用于RNA-seq分析的工具很多。不同工具采用的算法和模型各异,导致结果存在差异。
2017年,Nature Communication上发表的一篇论文详细比较了RNA-seq各种工具的性能,最终得出:DESeq2是最靠谱的分析工具之一。
生信人公众号解读:必看|史上最全的39个RNA-seq分析工具
推荐使用!
靠谱的基因富集分析工具:ClusterProfiler
同样的,基因富集分析的工具也比较多,这里推荐ClusterProfiler,原因有三:
- 支持人、动物、植物和微生物等各物种
- 软件仍然还在维护中
- 分析数据库实时更新
- 内嵌了丰富的可视化工具,随便修改一下就是发表级
推荐使用!
作者的微信公众号:biobabble ClusterProfiler应用文章目录点这里